Blog
October 28, 2025
Condensed Guide to Medical Device Requirements Management

Security & Compliance,
Application Lifecycle Management
With evolving global regulations, rising patient expectations, and rapid technological innovation, medical device manufacturers need development workflows that prioritize precision, agility, and compliance. This means gathering, specifying, and managing all requirements in an Application Lifecycle Management (ALM) tool that allows you to trace every requirement from origin to testing to release.
This guide provides a snapshot of current medical device compliance landscape and explains how to successfully engineer your requirements. We’ll explore the latest regulatory frameworks, the challenges they create, and how modern tools like Perforce ALM can help your teams bring products to market faster without risking quality or compliance.
Back to topMeeting Medical Device Compliance in 2026 and Beyond
If you’re in the medical device or medical software industry, you know that rules and standards continually evolve. In the coming year, we’ll see several significant changes that could impact how you develop and produce your products:
FDA (U.S.)
The FDA’s new Quality Management System Regulation (QMSR), effective February 2026, aligns U.S. requirements with ISO 13485:2016—the international standard for Quality Management Systems (QMS). This shift simplifies global compliance but requires updates to documentation, terminology, and internal processes.
The FDA also continues to refine guidance for Software as a Medical Device (SaMD) and AI-powered devices. The updated guidelines emphasize full algorithm transparency, continuous risk management, and post-market monitoring for AI-driven and software devices.
Additionally, the FDA has increased its fees for device registration and approval.
EU MDR (Europe)
The EU Medical Device Regulation (MDR) will enforce stricter classification rules for software. They will require robust clinical evidence, technical documentation, and post-market surveillance.
The In Vitro Diagnostic Regulation (IVDR) is now enforced, with ongoing extensions for select product groups.
SaMD developers must navigate Rule 11, which governs software used for diagnosis or therapy.
Cybersecurity
Increased threats to private data have elevated the need for stronger cybersecurity measures for all networked devices. Failure to maintain strong protections and comply with HIPAA and FDA/EU guidelines can result in regulatory action and loss of market confidence.
Summary of Key Regulatory Changes for Medical Devices
| Area | Key Change/Action | Effective Date | Impact on Requirements Management |
| FDA QMSR | 21 CFR Part 820 replaced by ISO 13485:2016-based QMSR | Feb 2, 2026 | Requires migration to globally standardized documentation and detailed tracking. |
| FDA AI Device Guidance | New requirements for post-market surveillance, transparency, and monitoring | Throughout 2025 | Adds mandatory ongoing algorithm performance tracking, detailed risk documentation, and transparency in requirements. |
| Cybersecurity | Heightened requirements and protocols for device data security | Ongoing | Introduces new technical and process requirements for secure design, threat monitoring, and cybersecurity measures. |
| FDA User Fees | Updated fee schedule, affecting registration and submissions | Oct 2024–Sept 2025 | Demands earlier budgeting and strategic planning for regulatory submissions. May affect how requirements are prioritized and phased. |
| EU MDR/IVDR Deadlines | Full MDR/IVDR compliance, notified body involvement, EUDAMED deadline | May 2025–Jan 2026 | Centralizes documentation in EUDAMED, increases traceability (UDI codes), demands ongoing clinical and risk evaluation, and stricter design controls. |
| EU Joint Assessments | EU-wide joint clinical assessments for select high-risk medical devices | Jan 2026 | Requires robust, harmonized clinical and safety data, cross-EU documentation, and enhanced post-market requirements. |
Accelerated Pathways
The net result of these changes is more pressure on manufacturers to maintain regulatory oversight and meet compliance. This can impact production schedules and increase development costs. To address these concerns, both U.S. and EU authorities are expanding accelerated pathways for breakthrough and innovative devices, but the process requires harmonized standards, increased evidence pooling, and comprehensive post-market surveillance. All manufacturers must prepare for significant operational and documentation shifts to maintain U.S. and European market access.
The Evolving Role of AI and IoT in Medical Devices
Another consideration is the integration of Artificial Intelligence (AI) and the Internet of Things (IoT) into medical devices. These technologies introduce new functionalities and, consequently, new types of requirements and risks.
The FDA’s guidance on Predetermined Change Control Plans (PCCPs) allows manufacturers to pre-authorize certain AI updates. This streamlines post-market changes if the outlined protocols and acceptance criteria are strictly followed. The EU’s AI Act classifies AI-driven medical devices as “high-risk” and introduces additional oversight that requires transparency and ethical safeguards.
- AI in Medical Devices: Devices with machine learning algorithms (AI/ML) can adapt and change their behavior based on new data. This requires a unique approach to requirements management, focusing on defining the performance boundaries of the algorithm, managing the data used for training and validation, and ensuring the device's "locked" algorithm remains safe and effective post-deployment.
- Example: GE’s SIGNA Champion MRI system utilizes advanced AI and deep learning technology to enhance its MRI scans. It benefits from FDA-cleared updates via a PCCP.
- IoT and Connectivity: Connected medical devices like wearables, smart beds, and infusion pumps transmit data over networks but introduce cybersecurity risks. Requirements must now cover data encryption, secure authentication, and protection against unauthorized access. The FDA has issued specific guidance on cybersecurity to manage risk in connected devices.
- Example: The Dexcom G7 glucose monitoring device offers users continuous glucose data via smartphone connection. As a result, the company needed to submit cybersecurity risk documentation to the FDA.
Managing these complex software-driven features demands a tool that can handle intricate software requirements, link them to hardware specifications, and trace them through risk mitigation and testing.
Back to top7 Best Practices for Managing Medical Device Requirements
A proactive requirements management strategy sets the foundation for regulatory success:
- Start with clear stakeholder input to ensure requirements are thorough and actionable.
- Align requirements with risk assessments, cybersecurity, and usability engineering to avoid costly surprises.
- Streamline requirements capture and approval with a documented process that integrates both hardware and software needs.
- Automate traceability using a dedicated ALM tool designed for medical device development that links every requirement to development, testing, and validation activities.
- Enable real-time change management so any updates are tracked, auditable, and easy to align with evolving regulations.
- Conduct regular reviews and audits to ensure all tests and issues are properly managed and resolved.
- Plan for post-market surveillance and updates to monitor device performance, user feedback, and adverse events.
Hazard Analysis and Risk Management in Medical Devices
From wearable monitors to surgical robots, product failure can lead to severe injury or death. To keep patients safe, you need a systematic approach to analyzing and managing risk.
ISO 14971:2019 is the international standard for medical device risk management. Adhering to this standard ensures compliance and helps prioritize development efforts effectively. By identifying potential hazards early, you can avoid late-stage design changes and costly recalls.
7 Key Steps in Medical Device Risk Management
- Define Acceptance Criteria: Establish what level of risk is acceptable. This is often documented in a risk matrix.
- Conduct Hazard Analysis: Identify all potential hazards related to the device's intended use.
- Perform Risk Analysis: Calculate a Risk Priority Number (RPN) for each identified risk (more below).
- Implement Risk Mitigation: Design and implement controls for unacceptable risks.
- Analyze Residual Risk: Evaluate the risk level after mitigation measures are in place.
- Determine Overall Risk Acceptability: Decide if the device's overall residual risk is acceptable.
- Monitor Post-Market: Continuously analyze and update risk assessments based on real-world data.
How to Calculate a Risk Priority Number
Risk analysis involves rating potential hazards based on their Severity (S), Occurrence (O), and sometimes Detection (D). These factors are used to calculate the RPN (S x O = RPN, or S x O x D = RPN), which helps prioritize which risks require immediate attention.
- Severity: Is a number 1-5 (with five being death) that defines how serious a potential failure would be.
- Occurrence: Describes how likely a failure is to happen.
- Detection: Indicates how easily a failure can be detected before it causes harm. Electrical shock, for example, is easily detected while detecting bacteria in a sterile environment is much more difficult (and would receive a higher rating).
It’s important to calculate a risk priority number that takes at least two factors into account because something that has a high severity but rarely occurs will take less priority than something with high severity that is difficult to detect.
Creating a FMEA or Risk Matrix
Traditional risk management methods like Failure Mode and Effects Analysis (FMEA) and risk matrices have long been used to identify and prioritize potential hazards. However, manual processes like spreadsheets often leave gaps in visibility, traceability, and consistency.
A better practice is to use a dedicated requirements and test case management tool such as Perforce ALM, that allows you to link failure modes to design inputs, requirements, and test cases. Perforce ALM can auto-populate your matrix with you RPN and adjusted RPN so that you have clear, comprehensive visibility into your entire risk management process.
Here is an example of a FMEA created by Perforce ALM, which visually details risk severity, occurrence, and detection to help teams prioritize their mitigation efforts:
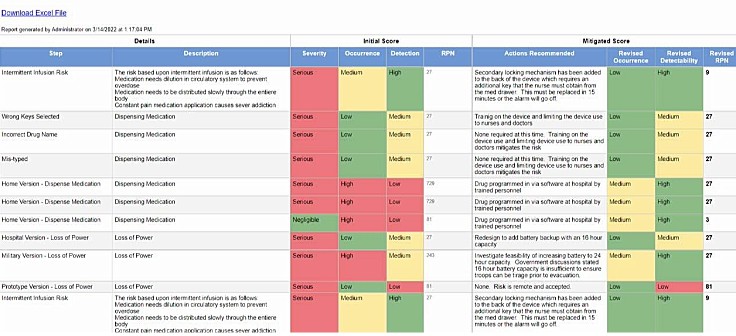
Additional Resource: Risk Management: Scoring that Matters to Product Developers. This blog identifies the types of risk and explains risk assessment in greater detail.
Back to topThe Top Challenges in Meeting Medical Device Requirements and How Perforce ALM Solves Them
1. Regulatory Complexity Across Global Markets
- Challenge: Navigating evolving regulations like FDA QMSR, EU MDR, and ISO standards increases the risk of delays and noncompliance.
- Solution: Use Perforce ALM to map requirements to specific frameworks and automate traceability across regions.
2. Innovation vs. Compliance (AI/ML Devices)
- Challenge: AI-powered devices face unclear classification and validation rules, especially under the EU AI Act.
- Solution: Perforce ALM supports version control and traceability for algorithm changes, enabling documentation of model updates and validation results.
3. Quality Management System (QMS) Overhaul
- Challenge: Transitioning to FDA’s QMSR and aligning with ISO 13485 requires retraining and revalidation.
- Solution: ALM automates documentation workflows and enforces standard operating procedure (SOP) adherence with minimal manual effort.
4. Cybersecurity & Data Integrity
- Challenge: IoT-connected devices are vulnerable to breaches and require robust cybersecurity and data transparency.
- Solution: ALM links security requirements to test cases and risk controls for compliance with FDA and EU MDR.
Additional Benefits of Perforce ALM for Medical Devices
- Full traceability across all artifacts
- Configures to your existing workflow
- Custom fields and filters
- Granular security permissions
- Provides a single source of truth across all documents
- Scalable collaboration across dispersed teams
- Affordable for small to medium teams
- Robust support options
Discover the Complete Medical Device Requirements Solution
Talk to a Perforce expert today or watch our free demo. You’ll see why Perforce ALM is the industry choice for managing medical device requirements securely, efficiently, and transparently throughout your development cycles.